
Newsroom





Mitochondrial DNA (mtDNA) mutations and depletion are significant contributors to mitochondrial epilepsy and neurodegeneration. However, studies to understand the mechanisms behind these issues and to develop therapies are limited due to the lack of suitable models. While caloric restriction (CR) is known to have neuroprotective effects in general, its impact on conditions involving mitochondrial dysfunction remains unclear.
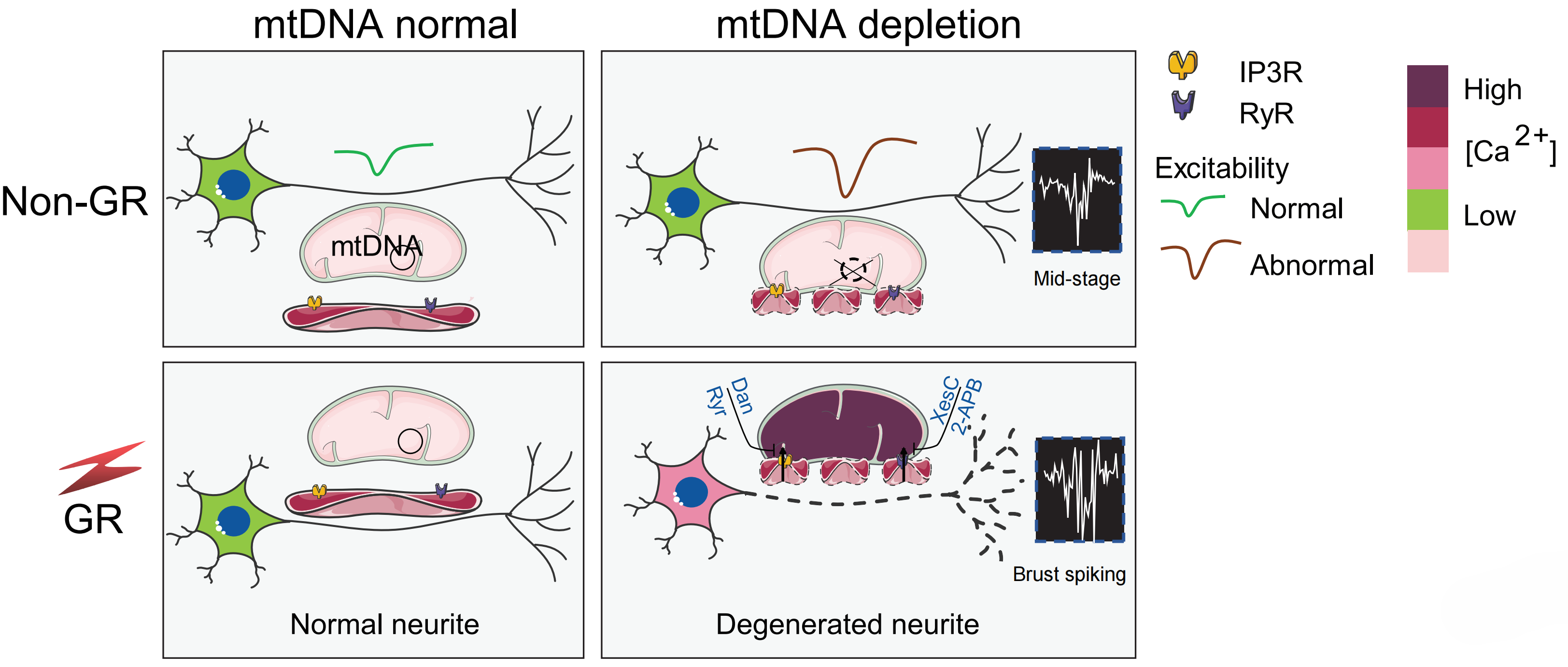
To tackle this issue, a research team led by Prof. LIU Xingguo from the Guangzhou Institutes of Biomedicine and Health of the Chinese Academy of Sciences (CAS) conducted a study and discovered that CR induces neurodegeneration in neurons with mtDNA depletion by disrupting calcium transfer between the endoplasmic reticulum (ER) and mitochondria. This pathological calcium signaling leads to early motor dysfunction, accelerated epilepsy progression, and increased neuronal damage.
This study was published in the journal Molecular Psychiatry.
Using PHP.eB vectors to deliver the HSV-1 UL12.5 protein—which targets mitochondria to induce selective mtDNA depletion—the researchers generated a mouse model that exhibited hallmark features of epilepsy. Electroencephalogram (EEG) recordings showed increased epileptic discharges, and patch-clamp studies revealed neuronal hyperexcitability, characterized by reduced sodium and potassium currents and heightened excitatory synaptic activity.
The researchers observed that treating an mtDNA-depleted human iPSC-derived neuronal model with two-deoxyglucose (2-DG), a CR mimetic, induced significant neurodegeneration, synaptic loss, and axonal fragmentation. These effects occurred independently of mitochondrial membrane potential collapse or transport defects.
Through calcium imaging under conditions mimicking CR, they observed depletion of ER calcium stores and excessive mitochondrial calcium uptake, leading to intracellular calcium overload. The team found that chelating intracellular calcium rescued the neurons, whereas removing extracellular calcium had no effect, confirming that the calcium dysregulation originated intracellularly.
Additionally, the researchers demonstrated that pharmacological inhibition of ER calcium channels (using RyR and IP3R antagonists) prevented 2-DG-induced neurodegeneration. This finding indicated that excessive calcium transfer from the ER to mitochondria drives neurotoxicity. In in vivo experiments, the researchers observed that fasting exacerbated seizures and gliosis in mtDNA-depleted mice. However, these effects were reversed by the IP3R inhibitor 2-APB, suggesting potential therapeutic strategies targeting ER-mitochondria calcium signaling.
This work not only identifies a novel neurodegenerative pathway linked to mitochondrial dysfunction and metabolic stress but also challenges the assumption that caloric restriction universally benefits health. It emphasizes the need for caution when applying CR to cases of mitochondrial epilepsy and related disorders.
This study was conducted in collaboration with multiple research groups, including Jinan University, the Innovation Center for Regenerative Medicine and Health at the Hong Kong Institute of Innovation under CAS, Guangzhou Medical University, and the Chinese University of Hong Kong.
Caloric restriction induces neurodegeneration in mtDNA-depleted neurons by altering ER-mitochondria calcium transfer. (Image by Prof. LIU's team)
