Wild relatives play a vital role in the genetic improvement of crops. The process of intentionally introducing chromosomal segments from wild relatives into crops through hybridization is known as distant hybridization. Wheat is one of the major staple crops most receptive to the introgression of alien chromosomes.
Understanding how these wild-derived segments contribute to wheat agronomic performance has long been a central research focus. Numerous genes controlling important wheat traits have been identified as originating from wild relatives, and large-scale sequencing efforts have cataloged these introgressed fragments in the wheat genome. However, the regulatory patterns of gene expression within these fragments remain poorly understood.
A research group led by HE Fei from the Institute of Genetics and Developmental Biology (IGDB) of the Chinese Academy of Sciences, in collaboration with LING Hongqing's team from IGDB, developed a novel strategy to address this challenge. By utilizing publicly available pan-genome resources, the researchers constructed a reference gene set that incorporates sequences from Triticeae wild relatives. This approach enables more comprehensive quantification of transcripts originating from introgressed alien segments.
Their findings were published in Nature Communications on December 15.The researchers generated an expression quantitative trait locus (eQTL) map for genes derived from Triticeae relatives using seedling transcriptome data from a wheat germplasm population. Their analysis revealed that, once incorporated into the wheat genome, the expression of genes from wild relatives is generally suppressed. However, stress-responsive genes tend to exhibit more active expression.
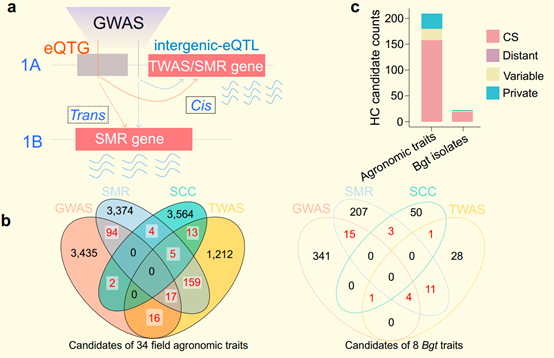
The researchers used the eQTL map to predict candidate genes for agronomic traits identified through genome-wide association studies (GWAS) by integrating multi-year, multi-location field trial data. These candidate genes are encoded not only by the standard "Chinese Spring" reference genome, but also by the genomes of diploid and tetraploid wild relatives. This provides a crucial foundation for gene cloning and functional characterization.
This study was supported by the National Key Research and Development Program of China, the National Natural Science Foundation of China, and the Yazhouwan National Laboratory.
Joint eQTLs of the pan-gene atlas and GWAS, TWAS, and SMR analysis of 34 field agronomic traits and 8 Bgt isolate infection phenotypes in wheat. (Image by IGDB)