
Protein interactions dominate almost all biochemical reactions in living cells, such as DNA synthesis, gene transcription and activation, protein translation, cell cycle regulation, and signal transduction. Through the study of these interactions, people have provided a new perspective to understand the occurrence and development of disease, thus providing molecular level information support for the prevention, diagnosis and treatment of specific diseases.
Due to limitations of experimental techniques, these methods are costly and time-consuming and have high false positive rates and false negative prediction rates when dealing with large-scale experiments. Therefore, the development of reliable computational methods has important practical significance in promoting protein-protein interaction identification.
Researchers from Xinjiang Technical Institutes of Physics and Chemistry of the Chinese Academy of Sciences developed Probabilistic Classification Vector Machines (PCVM), a computational method based on a PCVM model and zernike moments (ZM) descriptor for predicting the protein–protein interactions (PPIs) from protein amino acids sequences. The study was published in International Journal of Molecular Science.
Vectorization representation of amino acid sequences is a prerequisite for the application of machine learning methods to biological problems.
Researcher first used Position-Specific Scoring Matrix to represent each protein sequence, which retains the evolutionary information about proteins. And then, they employed the ZM method to generate feature vectors that be fed into classifier for implement prediction.
This method was performed on Yeast and Helicobacter pylori datasets with five-fold cross-validation experiments, which attained the average prediction accuracy of 94.48% and 91.25%, respectively.
The achieved AUC on Yeast was close to 97%. The results illustrate that the method can provide accurate, stable and high coverage prediction ability and provide a useful decision-making tool for genomics research.
Based on the above research results, the researchers realized a more accurate and stable prediction system by adding a deep learning system.
The experimental results display that this modified system can improve the accuracy of 2.2% and can realize cross species detection. The study was published in Molecular Biosystems.
This work was supported by the National Natural Science Foundation of China, National Science Foundation for excellent Young Scholars of China, and One Hundred Person Project of the Chinese Academy of Sciences.
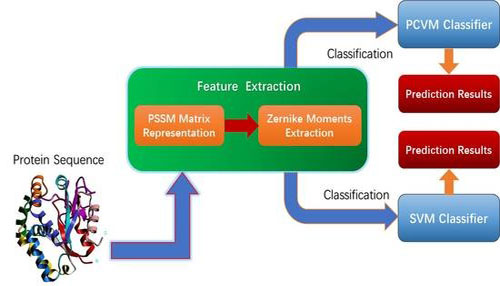
The flow chart of the PCVMZM (Image by XTIPC)

86-10-68597521 (day)
86-10-68597289 (night)

86-10-68511095 (day)
86-10-68512458 (night)

cas_en@cas.cn

52 Sanlihe Rd., Xicheng District,
Beijing, China (100864)
Copyright © 2002 - Chinese Academy of Sciences








